小ネタ:RDKit の update
新年、明けましておめでとうございます。
今年の目標として、技術力向上を掲げているので、きちんと勉強して積極的にブログを更新していこうと思います。
リハビリを兼ねて小ネタですが、RDKit の最新バージョンへの upgrade は conda で一発でできます。
conda update rdkit
調べれば一発で出てきますが、メモとして記載しておきます。
PySwarms による粒子群最適化
世の中はワクチンや東京オリンピックや AlphaFold2 で盛り上がっていますが、久々に書く記事は流行とは少し離れた内容です。
最適化手法というと、基本的な最急降下法や、それこそ流行りのベイズ最適化などがありますが、その一つに、粒子群最適化(partial swarm optimization: PSO)というものがあります。PSO は群知能(例えば渡り鳥一匹一匹はアホでも、集団で動くことで最適な行動を選択できることに相当する。コロナで最適な行動を取れない人類とは違う)の一種であり、1995 年に Kennedy と Eberhart により報告されています。
PSO では、グローバルな最適解を見出すのに、たくさんの点を用います(粒子群と呼ばれる所以です)。
各々の点は、普通の最急降下法のように、局所最適な方向に動こうとします。しかし、座標の更新にあたっては、局所的に最適な方向だけでなく、全ての点の中で最も良い座標も考慮します。そのため、PSO の最適化関数は、慣性項、ローカル項、グローバル項の 3 つの項から成り立っています。慣性項は、座標を更新(移動)する基本的なスピードを、ローカル項は、局所最適解を、グローバル項は、粒子群全体における最適値を考慮する役割をそれぞれ持っています(ざっくりとした理解なので、厳密な理解は原著論文をあたることをお勧めします)。
PSO ですが、Python で PySwarms というライブラリがあり、pip で簡単に入れることができます。
pip install pyswarms
PySwarm による PSO は動かすだけならかなり簡単に使えます。
import numpy as np import pandas as pd import pyswarms as ps # 最適化したい関数を定義(10, -3, 3 で最小値 0 になる) def object_function(x): y = (x[:, 0] - 10) ** 2 + (x[:, 1] + 3) ** 2 + (x[:, 1] + x[:, 2]) ** 2 return y # 最適化関数の重み(hyperparameter)を決める options = {"c1": 0.5, "c2": 0.3, "w":0.9} # 乱数固定 np.random.seed(0) # PSO を実行するインスタンスを設定する optimizer = ps.single.GlobalBestPSO(n_particles = 30, dimensions = 3, options = options) # Perform optimization cost, pos = optimizer.optimize(object_function, iters = 100) # 最適化された値を表示 print(cost) # 最適化された座標を表示 print(pos)
今回は 3 次元座標を入力すると数字が返ってくる関数を定義し、その最小値を探索することを考えます。
options で 3 つのパラメータを決めていますが、c1 がローカル項の重み、c2 がグローバル項の重み、w が慣性項の重みをそれぞれ示しています。w は 1 より大きくすると挙動がおかしくなるので、0~1 の範囲にしましょう。細かい範囲を探索するときは、値をオーダーレベルで小さくした方が良い気がします。c1 と c2 も重要なはずなのですが、筆者が変更した範囲では、あまり結果は変わりませんでした。
PySwarms は numpy に対応しているため、np.random.seed で乱数を固定し、結果を再現することができます(公式ドキュメントには書いていなかったが、明記しておいて欲しい)。
PySwarms では GlobalBestPSO と LocalBestPSO の二つのメソッドが使えますが、筆者が使ってみた限りでは差がありませんでした。
インスタンスを定義する際に、PSO の条件を色々設定できます。確実に使うのは、粒子数(n_particles)と dimension, option です。粒子数は、計算資源が許す限り、大きくしておいて損はないと思います。計算時間も、線形増加はしないです。dimension は、最適化したい目的関数のインプットの次元数です。今回は三次元データをインプットしているので、3 にしています。option では、PSO の最適化関数に関する設定を辞書として入力します。
optimizer.optimize で PSO を実行します。第一引数に最適化したい関数を入力します。関数は自分で定義できるので、機械学習モデルなどを用いることも可能です。また、値は小さくなるように更新されていくので、最大値を得たい場合は、関数にマイナスをつける必要があります。iters は繰り返しの回数で、これも単純に大きい方が良いですが、時間は線形増加します。
戻り値として、最適値(cost)と最適座標(pos)が帰ってきます。いずれも、最新の計算ではなく、今までの全ての計算の中で最適なものを算出しているので、計算のし過ぎによる悪影響はありません。
optimizer.cost_history で、最適値の履歴を時系列順のリストとして確認することができます。最適化が進んでいるか、プラトーに達しているかを判断できます。
optimizer.pos_history で、これまで計算した全ての座標を確認できます。時系列順のリストの中に、座標の numpy データが入っています。
状況によっては、初期座標を指定して計算したい場合もあると思います。座標を numpy 形式で用意し、optimizer を定義するときに init_pos で指定することができます。
# 初期座標を用意 initial_position = np.tile(np.arange(-15, 15) * 0.02 + 7, (3, 1)).T # init_pos で初期座標を設定 optimizer = ps.single.GlobalBestPSO(n_particles = 30, dimensions = 3, options = options, init_pos = initial_position)
探索範囲を指定したい場合は、optimizer の bounds で範囲を指定できます。最小値と最大値のタプルを用意します。タプルの次元数は、探索次元に合わせる必要があります。
# 最適化関数の重み(hyperparameter)を決める
options = {"c1": 0.5, "c2": 0.3, "w":0.9}
# 乱数固定
np.random.seed(0)
# 探索範囲を指定。一つ目のタプルは最小値を、二つ目のタプルは最大値を示す。
bounds = ((7, -5, 4), (9, 0, 6))
# init_pos で初期座標を設定
optimizer = ps.single.GlobalBestPSO(n_particles = 30, dimensions = 3, options = options, bounds = bounds)
# Perform optimization
cost, pos = optimizer.optimize(object_function, iters = 100)
# 最適化された値を表示
print(pos)
# 最適化された座標を表示
print(cost)最適化関数の重みである option は hyperparemeter ですが、gridsearch による最適化を PySwarms で行うことができます。
from pyswarms.utils.search import GridSearch # Grid search による最適化関数各項の重みの最適化 options = {"c1": [0.3, 0.5, 0.8], "c2": [0.3, 0.5, 0.8], "w": [0.2, 0.3, 0.5]} g_search = GridSearch(ps.single.GlobalBestPSO, n_particles = 10, dimensions = 3, options = options, objective_func = object_function, iters = 20) best_score, best_options = g_search.search()
PySwarms の基本的な使い方を今回はまとめてみました。PSO は現在でも時々使われている最適化手法なので、選択肢の一つに加えておくのも良いのではないかと思います。
Shapley 値によるモデルの解釈
以前の記事で、ランダムフォレスト(RF)による記述子の特徴量解釈について、少し触れました。
予測に関する記述子の役割をより深く考察するのに、shapley 値を用いた SHAP というものがあります。
Shapley 値はゲーム理論に基づき算出される値で、以下のブログに分かりやすく説明されています。
上のブログの例では、A、B、C の 3 人で力を合わせて 24 万円稼いだ場合、誰がどれくらい稼いだかを限界貢献度を元に表現しています。
A だけだと 6 万円、B だけだと 4 万円しか稼げませんが、A と B の二人だと 20 万円稼げます。ここで、B の仕事を A が手伝う(A が後から来る)とみなすと、A は 16 万円分働いていると考えられます。逆に、A の仕事を B が手伝う(B が後から来る)とみなすと、B は14 万円分働いたことになります。特徴量を追加する順番で稼いだ額、すなわち限界貢献度が異なりますので、順列を考え、全てのパターンの平均値を取ることで、Shapley 値が算出できます。
シンプルに考えるならば、Shapley 値が大きければ、目的変数に大きく寄与している、とみなせます。
SHAP を用いて機械学習モデルを解釈することもできます。
事前準備として、shap をインストールしておきましょう。
pip install shap
以前作成したLogS 予測 RFR モデルとデータを読み込んで、SHAP による解釈を行います。
import numpy as np import pandas as pd from rdkit import Chem from rdkit.Chem.Draw import IPythonConsole import shap import matplotlib.pyplot as plt import pickle # データの読み込み X_train = pd.read_csv("../data/LogS_train_scaled.csv", index_col = 0) X_test = pd.read_csv("../data/LogS_test_scaled.csv", index_col = 0) descriptor_label = X_train.columns y_train = pd.read_csv("../data/LogS_train_value.csv", index_col = 0) y_test = pd.read_csv("../data/LogS_test_value.csv", index_col = 0) # モデルの読み込み loaded_model = pickle.load(open("../model/RFR_LogS.sav", "rb")) # Shapley 値を計算 explainer = shap.TreeExplainer(model = loaded_model) train_explainer = explainer(X_train) test_explainer = explainer(X_test)
TreeExplainer で SHAP を実行するインスタンスを作成し、それにデータセットを入力することで Shaley 値を計算します。
実務上の注意点として、モデルに使用する特徴量の数が多いと SHAP の計算は時間がかかります。今回のモデルの構築には 94 記述子しか使っていないので、普通に計算できますが、数百記述子以上になると計算が中々終わらないので、気をつけて下さい。
train(test)_explainer はデータが格納されているインスタンスなので、色々取り出すことが可能です。例えば、 .values で Shapley 値そのものを取り出すことができます。
私が勉強不足なのでメソッドの詳細な説明は、公式ドキュメントや以下のブログに譲り、本記事では化学データの解釈に的を絞りたいと思います。
算出した Shapley 値を元に、データの解釈をしてみましょう。
まずは、bar plot を表示、出力してみましょう。
# 記述子の Shapley 値の平均値を bar plot として表示。 # 棒グラフに隣接している数値は Shapley 値であり、base value に加える値。 # 左の灰色の数字は元データにおける記述子の値 f = plt.figure(figsize = (8, 8)) shap.plots.bar(train_explainer[0]) # データを出力 f.savefig("../result/barplot_molecule.png", bbox_inches = "tight", dpi = 600)
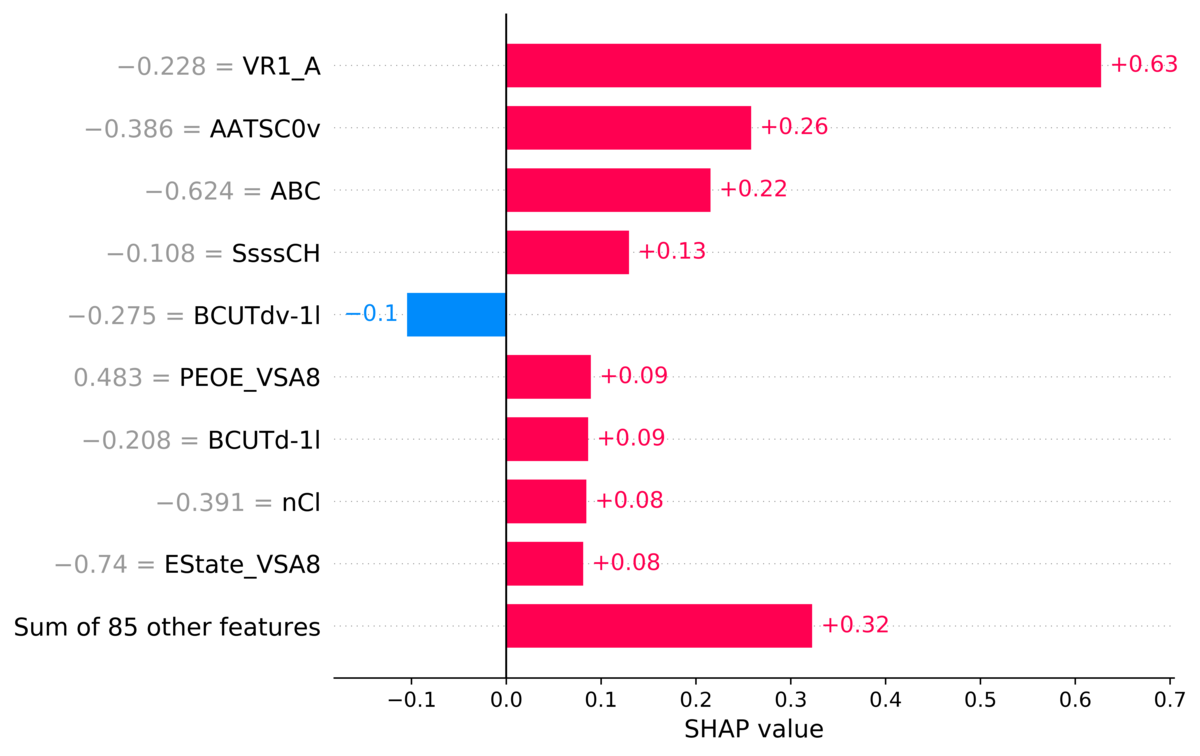
Bar plot では、各記述子についての Shapley 値の絶対値を示しています。Shapley 値はデータごとに計算されるので、feature importance のように、モデル固有の値でない点にも注意して下さい。
バーの真横にある赤 or 青で書かれているのが、あるデータ、記述子に対する Shapley 値です。赤だとプラス、すなわち予測結果の値を大きくするのに寄与し、青だとマイナス、すなわち予測結果の値を小さくするのに寄与します。
Base value が各モデル固有の値として与えられていますが、この Shapley 値を加えていくことで、予測結果が得られます。
ちなみに、左にある灰色の数字は、元の記述子の値です。
summary_plot を使っている記事が多いですが、bar で作ると色々融通が効きそうです。
細かい点を補足すると、explainer のインスタンスに対し、データの番号を指定することで特定のデータに対する bar plot を作れます。また、bar の関数の max_display という引数に数字を入力すると、plot で表示されるバーの本数を調整できます(一本は残りの記述子の Shapley 値を纏めたものになる点に注意)。bbox_inches を "tight" にしないと、プロットが見切れて出力されてしまう点も注意です。
Bar plot はモデル全体についても表示できます。
# モデル全体についての bar plot を表示 # 数字は絶対値の平均を表している点に注意。 f = plt.figure(figsize = (8, 8)) shap.plots.bar(train_explainer, max_display = 30) # データを出力 f.savefig("../result/barplot_whole.png", bbox_inches = "tight", dpi = 600)
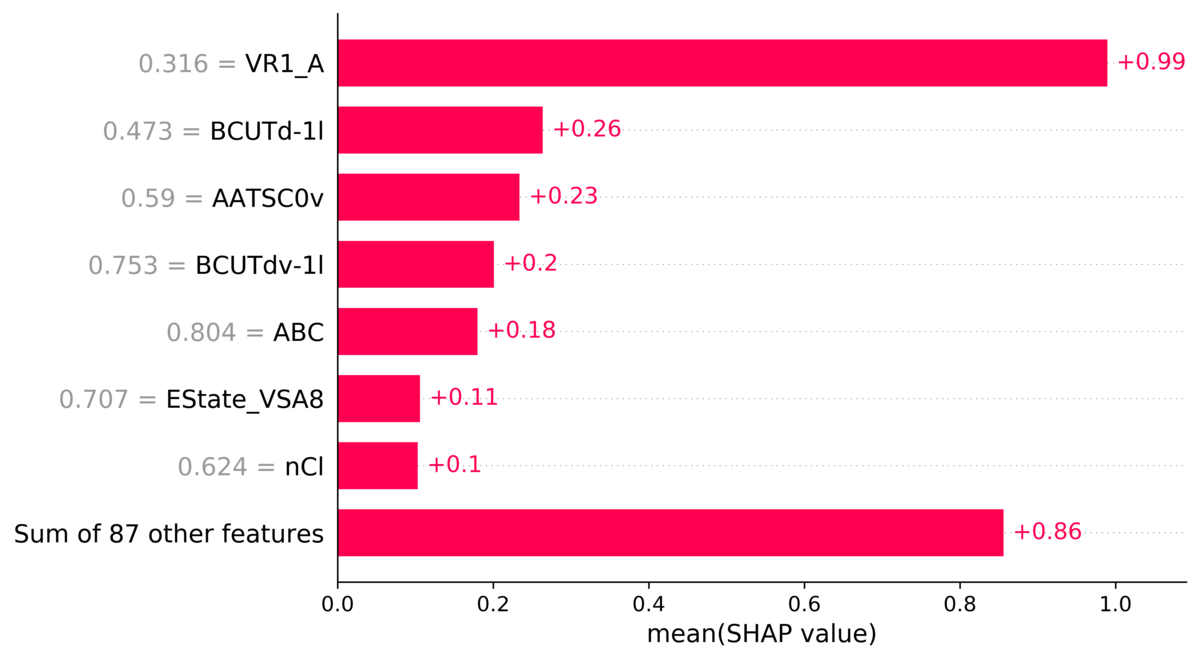
Shapley 値(と記述子の値)は、絶対値をとったものの平均値である点に注意してください。そのため、このプロットでは、値の増減にどう寄与したか判別することはできません。どの記述子が、予測値に大きく寄与したかは言えます。使い方としては、feature_importance に近いです(値や記述子の順番は一致するとは限りません)。
SHAP には他に force plot も用意されており、より視覚的にどの記述子が変化するかが見やすくなります。
# Force plot の表示 # 引数は base value, shapley 値のベクトル、記述子名 f = shap.plots.force(train_explainer.base_values[0], train_explainer.values[0], train_explainer.feature_names, matplotlib = True, show = False) # データを出力 f.savefig("../result/forceplot_molecule.png", dpi = 600)
Force plot も、全データを一度に表示させる機能はあるのですが、正直、あまり見やすいとは思わないので、説明は割愛します。
(他のプロットについては追記予定です)
その予測モデル、ちゃんと予測できてますか?〜applicability domain の考慮〜
機械学習モデルを研究や開発の現場で活用しようとする試みが盛んですが、様々な要因で難航しているのではないでしょうか。
その一つとして、折角苦労して作ったモデルが役に立たない、ということが挙げられます。機械学習モデルを作ることが目的ではなく、(論文になる、特許を取れる、臨床試験を突破できる、商品として売れるなどで)役に立つ化合物を作ることが目的であるはずです。機械学習モデルもあくまで道具です。金槌は釘を打つのには役に立っても、目玉焼きを作るのには役に立たないように、使い方を考える必要があります。
機械学習モデルの使い方を考察するのに有用な概念として、適用範囲(applicability domain: AD)が挙げられます。
AD はその予測モデルがうまく機能する範囲、というのが大まかな考え方です。例えば、ベンゼンやトルエン、アニリン、フェノール、ブロモベンゼンなど、一置換型ベンゼンのデータを集めて物性データ予測モデルを作り、予測に使うことを考えます。クロロベンゼンの物性を予測したい場合、元のデータ群と構造が似ているので、直感的に上手く予想できそうです。しかし、エストラジオールなどのステロイド化合物をこのモデルで予測しようとしても、構造が違い過ぎるので、上手く予測できなさそうです。
より細かくは、データ密度の話になるのですが、その辺りの詳細な説明は、明治大学の金子先生の HP にあります。
実務上重要となるのは、AD 外にあるデータは予測出来ない可能性がある、ということです。予測データが AD 外にあるかを予め確認できれば、予測結果の解釈や次の実験を計画するのに役に立ちそうです。
そうなると、どのようにして AD を判別するかが重要になりそうです。
AD を判別する簡便な方法として、k 最近傍法、one class support vector machine (OCSVM)、アンサンブル学習が考えられます。これも詳細は金子先生の HP にあります。
今回は、OCSVM を使って AD を予測してみます。
import numpy as np import pandas as pd from sklearn.svm import OneClassSVM from sklearn.decomposition import PCA from sklearn.linear_model import LinearRegression from sklearn.metrics import r2_score, mean_squared_error import matplotlib.pyplot as plt import pickle # データの読み込み X_train = pd.read_csv("../data/LogS_train_scaled.csv", index_col = 0) X_test = pd.read_csv("../data/LogS_test_scaled.csv", index_col = 0) # 記述子名のリストを取り出す descriptor_label = X_train.columns y_train = pd.read_csv("../data/LogS_train_value.csv", index_col = 0) y_test = pd.read_csv("../data/LogS_test_value.csv", index_col = 0) # One class support vector machine (OCSVM) モデルを構築する clf = OneClassSVM(nu = 0.3) y_train_class = clf.fit_predict(X_train) y_test_class = clf.predict(X_test) # 分割面からの距離を算出 clf.decision_function(X_test)
OCSVM は scikit-learn に含まれているので、その実装は簡単です。
与えるパラメータとして、nu があります。これは大雑把に言うと、トレーニングデータの中でAD 外とみなすデータの割合です。
目的やデータにより、任意に決める必要がありますが、肌感覚的には、0.2~0.3 辺りを試すと良い気がします。AD を厳しく見積もりたい時は、もっと値を大きくしても良いと思います。
OCSVM はラベルが必要ない、教師なし学習ですので、fit_predict (fit) には説明変数だけを入力すれば良いです。
y_train_class と y_test_class は各データについて 1 か -1 の値が書かれたベクトルとして得られます。1 であれば AD 内、-1 であれば AD 外となります。
decision_function を使うと、AD の境界となる分割面からの距離を算出することができます。正負の関係は上と同様です。
データの分布と AD の関係を、主成分分析(PCA)の第一主成分と第二種成分を取り出してプロットしてみます。
# データの分布を主成分分析(PCA)で表現してみる pca = PCA() X_train_pca = pca.fit_transform(X_train) X_test_pca = pca.transform(X_test) # フォントを変更 plt.rcParams["font.family"] = "Arial" # Plot のインスタンスと図の大きさを定義 fig = plt.figure(figsize = (10, 10)) # インスタンスの中に複数のグラフを作る場合に指定 # 引数は(行数、列数、具体的に作りたい図のインデックス) # グラフを 1 つだけ作りたいときは (1, 1, 1)としてやれば良い ax = fig.add_subplot(1, 1, 1) # グラフのタイトル ax.set_title("Applicability domain", fontsize = 28) # グラフの表示範囲 ax.set_xlim(-15, 15) ax.set_ylim(-15, 15) # 目盛の数字の大きさを調整 ax.tick_params(axis = "x", labelsize = 16) ax.tick_params(axis = "y", labelsize = 16) # 軸のラベルをつける ax.set_xlabel("PC_1", fontsize = 20) ax.set_ylabel("PC_2", fontsize = 20) # データをプロットする ax.scatter(X_train_pca[y_train_class == 1, 0], X_train_pca[y_train_class == 1, 1], c = "dodgerblue", label = "AD_inside") ax.scatter(X_train_pca[y_train_class == -1, 0], X_train_pca[y_train_class == -1, 1], c = "crimson", label = "AD_outside") # Legend をつける ax.legend(loc = "upper left", fontsize = 18) # 図を保存 fig.savefig("../result/AD_plot.png")
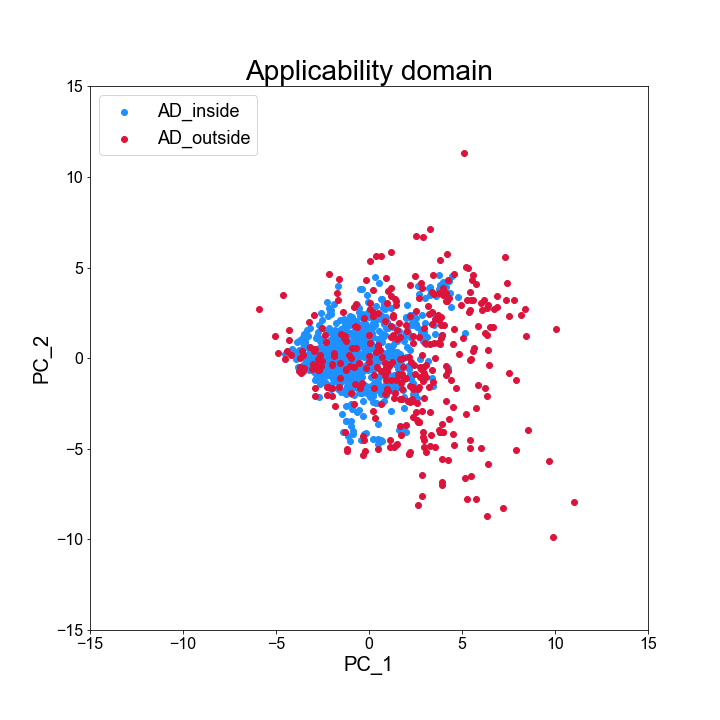
青(AD 内)は比較的原点に固まっているのに対し、赤(AD 外)は比較的原点から離れた領域に分散しているのが分かります。
続いて、テストデータについて、AD 内外で予測結果に影響するのか、y-y plot と R2 値を元に確認してみます。
# 線形重回帰モデル rgr = LinearRegression() rgr.fit(X_train, y_train) y_pred = rgr.predict(X_test) # y-y plot を作成 # フォントを変更 plt.rcParams["font.family"] = "Arial" # Plot のインスタンスと図の大きさを定義 fig = plt.figure(figsize = (10, 10)) # インスタンスの中に複数のグラフを作る場合に指定 # 引数は(行数、列数、具体的に作りたい図のインデックス) # グラフを 1 つだけ作りたいときは (1, 1, 1)としてやれば良い ax = fig.add_subplot(1, 1, 1) # グラフのタイトル ax.set_title("y-y plot", fontsize = 28) # グラフの表示範囲 ax.set_xlim(-9, 2) ax.set_ylim(-9, 2) # 目盛の数字の大きさを調整 ax.tick_params(axis = "x", labelsize = 16) ax.tick_params(axis = "y", labelsize = 16) # 軸のラベルをつける ax.set_xlabel("True_LogS", fontsize = 20) ax.set_ylabel("Pred_LogS", fontsize = 20) # 対角線(y = x)を引く ax.plot(list(range(-9, 3)), list(range(-9, 3)), "k") # データをプロットする ax.scatter(y_test, y_pred, c = "dodgerblue") # データをプロットする ax.scatter(y_test[y_test_class == 1], y_pred[y_test_class == 1], c = "dodgerblue", label = "AD_inside") ax.scatter(y_test[y_test_class == -1], y_pred[y_test_class == -1], c = "crimson", label = "AD_outside") # 図を保存 fig.savefig("../result/y_y_plot_withAD.png") # AD 内のデータについて R2 値を算出 print(r2_score(y_test[y_test_class == 1], y_pred[y_test_class == 1])) # AD 外のデータについて R2 値を算出 print(r2_score(y_test[y_test_class == -1], y_pred[y_test_class == -1]))
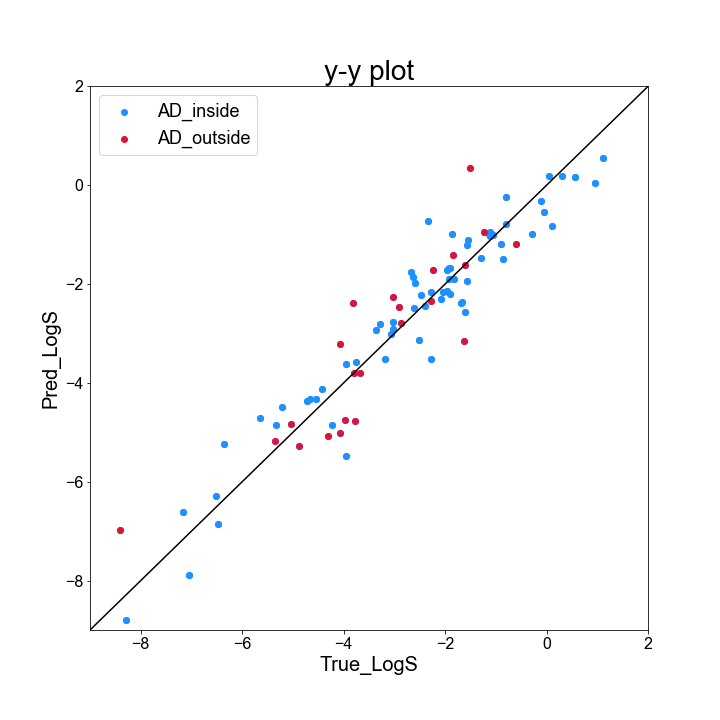
プロットを見ると、気持ち赤のプロットは対角線から離れているように見えますが、正直よく分かりません・・・
R2 値を見てみると、AD 内のデータが 0.920、AD 外のデータが 0.760 と、そこそこ差のある結果となり、AD 外のデータの予測が難しいことが見て取れます。
最後に、AD 内外の化学構造を確認してみます(コードはデータ構造がきれいではないので、解説はしないです)。
# テストデータに含まれる化学構造を表示 from rdkit.Chem.Draw import MolsToGridImage train_data_indices = pd.read_csv("../data/train_origin_tag.csv", index_col = 0) test_data_indices = pd.read_csv("../data/test_origin_tag.csv", index_col = 0) structures = Chem.SDMolSupplier("../data/logSdataset1290_2d.sdf") # AD 内に含まれるデータの構造を選択 test_structure_indices = [i.item() for i in np.array(test_data_indices)[y_test_class == -1][:20]] test_structure_examples = [structures[i] for i in test_structure_indices] # PNG 画像を作成し保存 img = MolsToGridImage(test_structure_examples, molsPerRow = 5, subImgSize = (300,200)) img.save("../result/test_outsideAD.png")
AD 外に含まれる構造は、AD 内に含まれる構造よりも複雑で、外れ値となりそうなのが化学的にも見て取れると思います。
AD を考慮することで、より適切な機械学習モデルの運用が実現すると思います。
小ネタ:Mordred で算出した記述子の欠損値処理
ケモインフォマティクスにおける Mordred は、化学構造から簡便に記述子を算出してくれる便利なライブラリです。
しかし、どんな分子でも記述子を計算してくれる訳ではなく、時々計算が出来ずにエラーを吐き出します。
エラーの種類は色々あるようですが、化学グラフの処理なんかでエラーが出るようです(メタンはエラーとなる記述子が多い)。
例えば mordred.error.Missing at 0xa1f90a510 といった具合です。
厄介なのが、エラーはそれ自体がオブジェクトとして dataframe のセルに入力されるため、 pandas 上では欠損値とみなされません(例えば dataframe.isnull() で検出できない)。そこで、コード上の処理に工夫が必要となります。
一番簡単なのは、数字以外が書かれている記述子を除いてしまうことでしょう。Pandas には select_dtype というコマンドがあり、指定した型のデータだけを抽出してくれるのですが、引数に "number" と指定することで、 int 型と float 型、つまり数字だけを選択してくれます。
import numpy as np import pandas as pd from rdkit import Chem from rdkit.Chem.Draw import IPythonConsole from mordred import Calculator, descriptors # 記述子計算用インスタンスを用意 calc = Calculator(descriptors, ignore_3D = True) # 計算したい化合物データを用意(mol 形式のリストを作る) mols = [Chem.MolFromSmiles(mol) for mol in ["C", "c1ccccc1", "CCI"]] # Pandas のデータフレームとして出力 df = calc.pandas(mols) # 数字以外が記載されている列を落とす df.select_dtypes("number")
select_dtype は、通常の解析においても力を発揮してくれるので、覚えておいて損はないと思います。
また、mordred は上記の設定だと、1613 記述子を計算してくれるのですが、そもそも特定の記述子だけが欲しいという場面もあります。既にモデルがある場合は、そのモデルで使っている記述子だけを計算すれば充分です。
その方法は、こちらの qiita の記事に纏まっています。
Qiita のコードとほぼ同じですが、下に例を記載します。
# 計算したい記述子名をリスト形式で用意 my_desc_names = ["ABC", "MW"] my_descs = [] # 一度計算するインスタンスを作成し、実際に計算するオブジェクトを一つ一つ取り出して確認する calc_dummy = Calculator(descriptors, ignore_3D=False) for i, desc in enumerate(calc_dummy.descriptors): if desc.__str__() in my_desc_names: my_descs.append(desc) # 後は通常通りに計算 calc = Calculator(my_descs, ignore_3D = True) mols = [Chem.MolFromSmiles(mol) for mol in ["C", "c1ccccc1", "CCI"]] df = calc.pandas(mols)
では、選択した記述子を使いたいにも関わらず、欠損値が含まれている場合はどうすれば良いでしょうか?
良い解決策が思いつかなかったので、下のように力押しをしています。
# 取り敢えず numpy 形式にする A = np.array(df) # 力押しで全てのセルをチェック for row_idx in list(range(A.shape[0])): for column_idx in list(range(A.shape[1])): # 数字でなければ float にできないので、nan になる if np.isnan(np.float(A[row_idx, column_idx])): # 欠損値を取り敢えず 0 で埋める A[row_idx, column_idx] = 0
あるいは、欠損値が含まれる記述子だけを選択してチェックをしていっても良いかもしれません。
# 欠損値が含まれている記述子を選択 lack_descriptors = [i for i in df.columns if i not in df.select_dtypes("number").columns] # 力押しで上で選ばれた記述子のセルを全てチェック for descriptor_name in lack_descriptors: for row_idx in list(range(df.shape[0])): # 欠損値を取り敢えず 0 で埋める df[descriptor_name][row_idx] = 0
無理に欠損値を使いたい場合は、ある程度限られてくると思うので、多少力押しになっても、実用上何とか耐えられる場合もあると思います。
いずれにしても、mordred そのもので何とかするというよりは、pandas などの力に頼った方が早いと思います。
小ネタ:ランダムフォレストの実装 + モデルの保存方法 + プロット作成の基本
ケモインフォマティクスでは回帰問題、あるいは判別問題をよく扱います。様々な機械学習モデルがあると思いますが、最も使いやすいモデルとして、ランダムフォレスト(RF)があります。RF の数学的原理を解説しているサイトは多数あるので、ここでは触れません。
RF を使う実務上のメリットとして、以下の点が挙げられます。
・特徴量の前処理をしなくても良好なモデルが得やすい。
・どのような特徴量が予測に有用なのか、寄与率(feature importance)を基に考察ができる。
・パラメーターチューニングをそこまでしなくても良好なモデルが得やすい(n_estimator (木の本数)を取り敢えず増やしておけば良い)。
・並列化が用意(n_jpbs という引数がある)
・データセットのサイズが小さくてもモデルが組める。データサイズが数千ぐらいだと、gradient boosting より性能が良いこともしばしば。
外挿に弱いなどの欠点もありますが、基本的には初心者でも扱いやすく、かつ良い結果が出やすいので、個人的には非常にオススメする手法です。
何かデータがあったときは、取り敢えず RF を試してみると良いと思います。
以下、モデルの解釈、保存まで含めた実装例です。
import numpy as np import pandas as pd from sklearn.ensemble import RandomForestRegressor from sklearn.metrics import r2_score, mean_squared_error import matplotlib.pyplot as plt import pickle # データの読み込み X_train = pd.read_csv("../data/LogS_train_scaled.csv", index_col = 0) X_test = pd.read_csv("../data/LogS_test_scaled.csv", index_col = 0) # 記述子名のリストを取り出す descriptor_label = X_train.columns y_train = pd.read_csv("../data/LogS_train_value.csv", index_col = 0) y_test = pd.read_csv("../data/LogS_test_value.csv", index_col = 0) # モデルの構築 rgr = RandomForestRegressor(random_state = 0) rgr.fit(X_train, y_train) y_pred = rgr.predict(X_test) # R2 値を表示 print("R2:") print(r2_score(y_test, y_pred)) print("-----\n") # 平均二乗誤差(RMSE)を表示 print("RMSE:") print(mean_squared_error(y_test, y_pred)) # モデルを保存 pickle.dump(rgr, open("../model/RFR_LogS.sav", "wb"))
今回のデータは、こちらのコードで作成したものを読み込んでいます。
データを読み込んだ時点では pandas の dataframe の形式になっているので、columns で記述子名を取り出します。これは、記述子の寄与率解釈の時に使います。
RF の実装は 3 行で出来てしまいます。最初にインスタンスを作成し、fit で学習データを入力、predict で予測ができます。前処理がいらないので、これでモデルの完成です。木の本数を増やしたいときは、最初のインスタンス作成の時点で n_estimators に数字を指定すれば良いです。取り敢えず大きい数にすれば予測性能は上がりますが、一定の数で頭打ちになるのと、本数が多いと計算時間がかかるので、マシンスペックと相談して決めると良いです。
ちなみに、判別モデルのときは、あるクラスに分類されると予測した木の本数の割合を、そのクラスに分類される確率とみなすことができます。この確率は、predict の代わりに predict_proba を指定することで算出することができます。
R2 と RMSE の表示もしてみます。R2 の説明は、上に出した記事に書いてあるので、省略します。RMSE は、どれくらい予測結果に誤差があるかを示す指標です。負ではない値を取り、0 に近い程、誤差が小さいので良好な予測が出来ていると解釈します。扱うデータや他のモデルとの比較によって、数字の良し悪しが変わるので、絶対値だけで解釈しないよう、注意が必要です。
モデルの保存は pickle.dump を使います。モデルを書き込むので、 open の引数に "wb" を指定します。覚えていれば一瞬なのですが、書く時に忘れがちなので、自分のメモとして記載しておきます。
モデルを保存したら、読み込みたい場面も出てきます。pickle.load で作成した機械学習モデルを読み込めます。気になる人は、以下のコードを試してみて、ちゃんと結果が再現できるか試してみて下さい。
# モデルの読み込み loaded_model = pickle.load(open("../model/RFR_LogS.sav", "rb")) # 予測結果が同じか確認 y_loaded_pred = loaded_model.predict(X_test) # R2 値を表示 print("R2:") print(r2_score(y_test, y_loaded_pred)) print("-----\n") # 平均二乗誤差(RMSE)を表示 print("RMSE:") print(mean_squared_error(y_test, y_loaded_pred))
寄与率を基に、どのような記述子が予測に有用か、確認をしてみましょう。
# 寄与率が高い特徴量を表示 feature_importances_top_indices = np.argsort(loaded_model.feature_importances_)[::-1][:15] for i in feature_importances_top_indices: print(descriptor_label[i]) print(loaded_model.feature_importances_[i]) print("----------\n")
RF モデルのインスタンスに feature_importances_ とあるので、これで記述子の特徴量を表示出来ます。ただし、学習に用いた記述子の順番通りに並んでおり、どの記述子が一番重要なのか分かりにくいです。そこで、記述子の大きい順に並び替えをしてあげて、上位何種類か(今回は 15 種類)をインデックスとして取得します。このインデックスを基に、特徴量の大きい順に値と記述子名を表示していきます(ここで最初に取得した記述子ラベルを使います)。
最後に、実測値と予測値がどの程度一致しているか、y-y plot を基に確認してみましょう。y-y plot は、実測値と予測値を二次元上にプロットした図であり、対角線(実測値と予測値が一致)に近い程良いと解釈する図です。Python だと、matplotlib を使うと綺麗な図が書けるのですが、少し扱いが難しいです。y-y plot 作成コードの例は以下の通りです。
# y-y plot を作成 # フォントを変更 plt.rcParams["font.family"] = "Arial" # Plot のインスタンスと図の大きさを定義 fig = plt.figure(figsize = (10, 10)) # インスタンスの中に複数のグラフを作る場合に指定 # 引数は(行数、列数、具体的に作りたい図のインデックス) # グラフを 1 つだけ作りたいときは (1, 1, 1)としてやれば良い ax = fig.add_subplot(1, 1, 1) # グラフのタイトル ax.set_title("y-y plot", fontsize = 28) # グラフの表示範囲 ax.set_xlim(-9, 2) ax.set_ylim(-9, 2) # 目盛の数字の大きさを調整 ax.tick_params(axis = "x", labelsize = 16) ax.tick_params(axis = "y", labelsize = 16) # 軸の名称を指定 ax.set_xlabel("True_LogS", fontsize = 20) ax.set_ylabel("Pred_LogS", fontsize = 20) # 対角線(y = x)を引く ax.plot(list(range(-9, 3)), list(range(-9, 3)), "k") # データをプロットする ax.scatter(y_test, y_loaded_pred, c = "dodgerblue") # 図を保存 fig.savefig("../result/y_y_plot.png")
最初に、図のフォントを指定します。細かい設定をするには他の操作が必要だと思いますが、今回は取り敢えず arial で統一してしまいます。次に、figure のインスタンスを作成します。ここで、図全体の大きさをインチ単位で指定します。そして、add_subplot で図のどこにグラフ (axes) を作成するか指定します。ここが分かりにくいのですが、figure で図全体を作成し、そこに各グラフ(axes インスタンス)を放り込んで細かく作っていく、というのが大雑把なイメージだと思います。また、matplotlib で図の作り方を調べると、plt に色々追加していくやり方も多く見られますが、図を細かく指定するには、axes インスタンスに対し操作をしていくやり方に慣れておいた方が良いと思います。Subplot を使って複数の図を作る事例が多く紹介されていますが、最初は一つの図だけを作る場面の方が多いと思うので、add_subplot は(1, 1, 1) で良いです。
グラフの表示範囲は、必ず全てのデータが範囲内に含まれているか確認して下さい。範囲外にデータがあると、プロットされません。
plot で線を、scatter で点を描くことができます。
プロットの色は、簡単な色であれば、一文字で表現できます ex) "k": 黒
また、以下の表にある色は文字で指定できます。
作成したプロットは、こんな感じです。
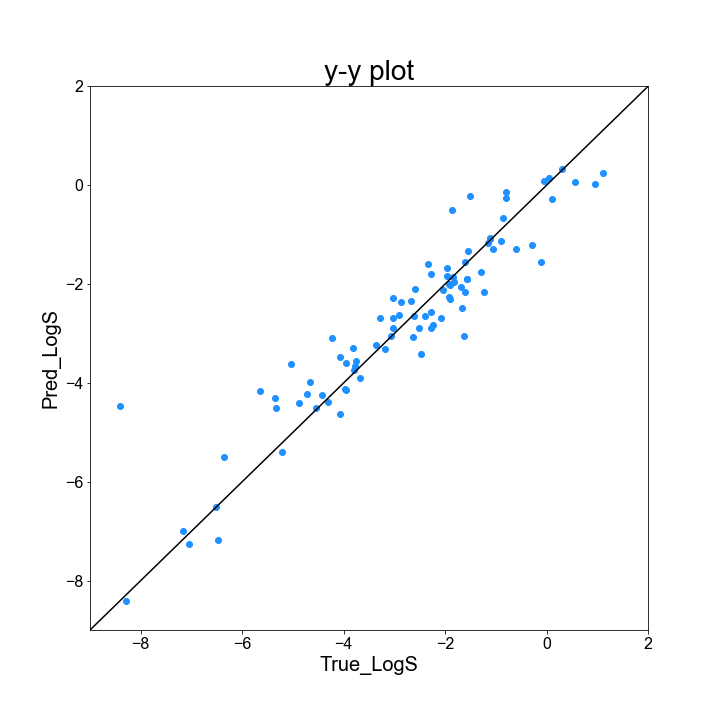
対角線の左上にあると、実測値よりも大きく、右下にあると実測値よりも小さく LogS を予測したことになります。
グラフィカルユーザーインターフェース(GUI)の作り方入門
ケモインフォマティクスと聞くと、機械学習的なアプローチを思い浮かべる人も多いかと思いますが、伝統的には、解析ツールを化学者に提供する、というのも重要な役割です。例えば Gaussian や GAMESS は有用な量子化学計算ソフトウェアですが、そのグラフィカルユーザーインターフェース(GUI)が使いやすいことも、広く普及した要因の一つだと思います。
論文で発表された解析用のツールなどはよく GitHub などに上がっているので、ケモインフォマティシャンなどはすぐに使える場合も多いですが、実験化学者の方々全員がプログラミングに精通しているわけではありません。解析ツールが現場で気軽に使われるためには、優れた GUI を提供する必要があります。
構造式とその分子量を表示する GUI を作ってみました。テキストボックスに SMILES を入力し、ボタンを押すと分子量と構造式が出る GUI です。
import numpy as np import pandas as pd from rdkit import Chem from rdkit.Chem.Descriptors import MolWt from rdkit.Chem.Draw import IPythonConsole, MolToImage import tkinter as tk from PIL import Image, ImageTk # 結果を表示するウィンドウの設定 def createNewWindow(): newWindow = tk.Toplevel(root) newWindow.title("Result") newWindow.geometry("700x700") # テキストボックスに入力した SMILES を取り出して分子量を表示 mol = Chem.MolFromSmiles(textbox.get()) result = MolWt(mol) weight = tk.Label(newWindow, text = str(result), foreground = "dodgerblue") weight.grid() # 構造式の表示 img = ImageTk.PhotoImage(MolToImage(mol)) # 構造式を表示する領域を指定 canvas = tk.Canvas(newWindow, bg = "black", width = 400, height = 400) canvas.place(x = 100, y = 100) canvas.create_image(200, 200, image = img, anchor = tk.CENTER) # 結果のウィンドウの表示 newWindow.mainloop() # メインのウィンドウの設定 root = tk.Tk() # ウィンドウ名 root.title("Molecule viewer") # ウィンドウのサイズ root.geometry("500x500") # ウィンドウ中の文章 label = tk.Label(root, text = "Input SMILES", foreground = "crimson") label.grid(row = 0, column = 1) # テキストボックスの設定 textbox = tk.Entry(root) textbox.insert(tk.END, "CCO") textbox.grid(row = 1, column = 1) # ボタンの設定 button = tk.Button(root, text = "Show", command = createNewWindow) button.grid(row = 1, column = 0) # 実行 root.mainloop()
GUI 作成用のライブラリとして、tkinter を用いました。tkinter はデフォルトで入っているので、別途インストールする必要はありません。
GUI ライブラリとしては、wxPython を本当は使ってみたかったのですが、別途インストールが必要なのと、Jupyter Notebook で使おうとすると、カーネルを設定する必要があったので、今回は使用を見送りました。
最初に、メインのウィンドウのインスタンスを tk.Tk() で作ります。ウィンドウの名前やサイズなどの設定を、このインスタンスに入力していくイメージです。
geometry で初期ウィンドウのサイズを決められます。縦x横を文字列として入力します。真ん中の x は普通に小文字のアルファベットの x です。
tk.Label() で、ウィンドウ内に文章を挿入できます。foreground で色を設定できますが、matplotlib と同様に、名前で指定することもできます。
テキストボックス(検索欄など、自由に文字を入力できる枠)は、tk.Entry() で設定できます。引数を何も設定しなくても今回は動きましたが、より複雑になるとエラーを起こしそうなので、インスタンスを指定した方が良い気がします。insert でデフォルトの入力を用意することもできますが、無くても問題ありません。tk.END はデフォルトの文字をどう枠内に置くか、の設定で、とりあえずは”おまじない”で良いです。grid でパーツをウィンドウ内のどの部分に置くかを設定できます。場所の設定関数は、grid、place、pack の 3 種類がありますが、grid と pack を併用したらエラーが出たので、統一した方がいい気がします。row と column で大体の位置を指定できますが、より細かく位置設定をしたい場合は、他の方法を使う必要がありそうです。
ボタンも tk.Button() を使って、テキストボックスと似た感じで設定できます。大切なのは command という引数で、ボタンを押すと、指定したオブジェクトを実行してくれます。今回は、新しい画面を出力する自作関数を指定しています。
結果のウィンドウは、メインのウィンドウとは別に用意します。tk.Toplevel(root) で新しいインスタンスを作ります。親のインスタンスを指定することで、対応関係を作ります。結果のウィンドウについても、メインと同様に要素を追加していきます。分子量は float 型として出てくるので、str 型に変換して、文字列として出力できるようにします。
構造式の表示は、mol オブジェクトを MolToImage(mol) で png に変換し、ImageTk.PhotoImage(png) で GUI で表示できるオブジェクトに変換します。
GUI 側は、tk.Canvas() で画像を表示できる領域を用意します。bg は background のことで、画像を貼り付ける領域の色です。プリントなどを貼り付けるボードの色、というのがイメージしやすいでしょうか。anchor で画像の位置を指定できますが、任意の位置に来るようにするには、canvas のパラメータも関連するため、少しコツが必要です。
mainloop() で GUI を表示することができます。逆に、Jupyter Notebook で変数などを確認したい場合は、mainloop() を除くとやりやすいです。mainloop() は各ウィンドウに対して用意します。
上記のコードを Jupyter Notebook で実行して出てきた画面は以下の通りです。結果は default のエタノールではなく、フェノールにしてあります。
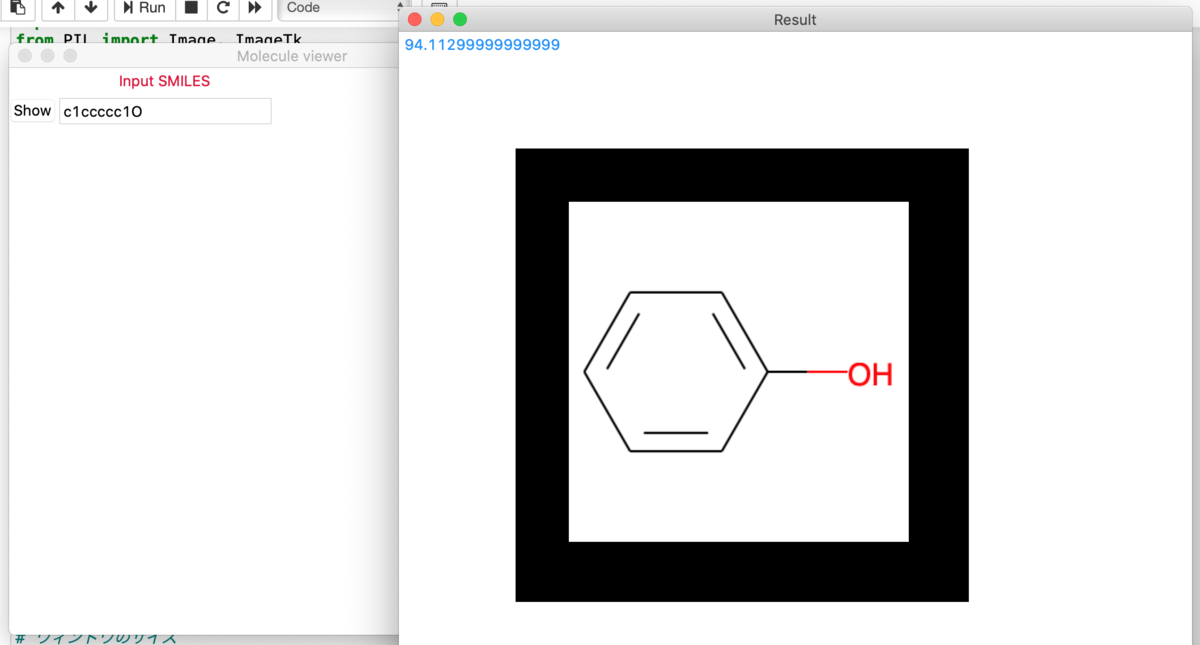
分子量(青字)と構造式が結果の画面に示されています。ウィンドウの左上には、ちゃんと拡大や終了ボタンもあります。
赤いボタンを押してメインのウィンドウを閉じれば、このコードも終了します。逆に、閉じないと、Jupyter Notebook ではこのコードのあるセルを実行し続けているので、他のことはできなくなってしまいます。
今回はとりあえず動くものを作ってみましたが、工夫次第で、多機能かつデザイン性にも優れた GUI を作ることもできそうです。
少しシステムデザインよりの話ですが、この辺りをやるケモインフォマティクスの人が増えてもいいのかな、とも思います。