RDKit の基本的な使い方
RDKit はケモインフォマティクスを行う上で、必要不可欠なツールです。
大体のことは RDKit で出来てしまうのですが、公式ドキュメントがめちゃくちゃわかりにくいという致命的な欠点があります。
私が使った範囲で、RDKit の機能についてメモを示します。
RDKit のインストール (pip)
pip install rdkit
RDKit のインストール (conda)
conda install -c rdkit rdkit
RDKit のバージョン確認
import rdkit
rdkit.__version__
SMILES から mol オブジェクトへの変換
from rdkit import Chem mol = Chem.MolFromSmiles("CCO")
Mol オブジェクトから SMILES への変換
smiles = Chem.MolToSmiles(mol)
Canonical SMILES への変換
canonical_smiles = Chem.MolToSmiles(Chem.MolFromSmiles("c1ccnc(Cl)c1"))
同じ要領で SMARTS、Inchi、CXSMILES 等も扱える
smarts = Chem.MolToSmarts(mol) inchi = Chem.MolToInchi(mol) cxsmiles = Chem.MolToCXSmiles(mol)
水素原子を付ける
mol_h = Chem.AddHs(mol)
隣接行列の算出
Chem.GetAdjacencyMatrix(mol)
距離行列の算出
Chem.GetDistanceMatrix(mol)
距離行列を用いて Wiener index も算出できる
np.sum(Chem.GetDistanceMatrix(mol)) / 2
分子量の算出
from rdkit.Chem.Descriptors import ExactMolWt ExactMolWt(mol)
QED の算出
from rdkit.Chem.Descriptors import qed qed(mol)
SA score の算出
import sys import os from rdkit.Chem import RDConfig sys.path.append(os.path.join(RDConfig.RDContribDir, "SA_Score")) import sascorer sascorer.calculateScore(Chem.MolFromSmiles("c1ccccc1OC(=O)O"))
TPSA の算出
from rdkit.Chem.rdMolDescriptors import CalcTPSA mol = Chem.MolFromSmiles("CCO") CalcTPSA(mol)
Labute ASA の算出
from rdkit.Chem.MolSurf import LabuteASA mol = Chem.MolFromSmiles("CCO") # 立体配座は不要だが、水素原子の有無で値が変わる。 mol = Chem.AddHs(mol) LabuteASA(mol)
水素原子の授受に関連する官能基
from rdkit.Chem.Lipinski import NumHDonors, NumHAcceptors mol = Chem.MolFromSmiles("CC(=O)c1c(O)cccc1") # 水素原子受容体の数 NumHAcceptors(mol) # 水素原子供与体の数 NumHDonors(mol)
分子中の全炭素原子のうち SP3 炭素の割合
from rdkit.Chem.Lipinski import FractionCSP3 mol = Chem.MolFromSmiles("FC(F)(F)c1cc(O)ccc1C=CC") FractionCSP3(mol)
分子中に含まれる環構造の数を算出。縮合環は別々に数える(例:ナフタレンは 2 とカウント)。
from rdkit.Chem.Lipinski import RingCount mol = Chem.MolFromSmiles("c1c2ccc(CCC3CC3)cc2cc(C(=O)O)c1") RingCount(mol)
立体異性体や幾何異性体を網羅的に発生させる(不斉中心や cis/trans の考慮)
from rdkit.Chem.EnumerateStereoisomers import EnumerateStereoisomers mol = Chem.MolFromSmiles("NC(C=CC)C(=O)O") [m for m in EnumerateStereoisomers(mol)]
指定した部分構造に対応する index を取得する
mol = Chem.MolFromSmiles("c1c(Cl)c(c2nccc(OCCC)c2)ccc1C(=O)NC1CC1") query = Chem.MolFromSmiles("CCC") mol.GetSubstructMatches(query)
分子中の scaffold を取得
from rdkit.Chem.Scaffolds.MurckoScaffold import GetScaffoldForMol, MakeScaffoldGeneric mol = Chem.MolFromSmiles("c1c(OC)c(Cl)ccc1NC(=O)C1CC1CC") # Bemis-Murcko scaffold の取得。環構造と linker とアミドのカルボニル酸素から Bemis-Murcko scaffold は構成される。 GetScaffoldForMol(mol) # 構造式中の原子を全て飽和炭素に置き換えた構造に変換する(グラフが同じアルカン)。 MakeScaffoldGeneric(mol)
共通する部分構造(Maximum Common Substructure: MCS)を抽出
from rdkit.Chem.rdFMCS import FindMCS mols = [Chem.MolFromSmiles(s) for s in ["c1ccccc1OC", "Oc1ccccc1C(=O)O", "Oc1cc(O)cc(O)c1"]] mcs = FindMCS(mols) Chem.MolFromSmarts(mcs.smartsString)
Mol オブジェクトから atom オブジェクトを取り出す。引数は mol オブジェクト内の原子の index。原子番号ではなく、mol オブジェクトによって index が異なる場合があるので注意。
mol.GetAtomWithIdx(0)
Mol オブジェクト中の atom オブジェクトを全て取り出す。
[atom for atom in mol.GetAtoms()]
Atom オブジェクトの質量(原子量)を取り出す。
atom.GetMass()
不斉点の情報(原子の index と R/S)を取得。
mol = Chem.MolFromSmiles("N[C@@H]([C@H](O)C)C(=O)O")
Chem.FindMolChiralCenters(mol)
Mulliken 電荷とグラフ構造から算出される部分電荷である Gasteiger charge を取得。
from rdkit.Chem.rdPartialCharges import ComputeGasteigerCharges # Gasteiger charge の算出 ComputeGasteigerCharges(mol) # Gasteiger charge は atom オブジェクトの property に str 型として記載されている。計算で用いたい場合は型を変換する。 atom = mol.GetAtomWithIdx(0) float(atom.GetProp("_GasteigerCharge"))
Mol オブジェクトから bond オブジェクトを取り出す。引数は mol オブジェクト内の結合の index。Atom オブジェクトと扱いが似ている。
mol.GetBondWithIdx(0)
Mol オブジェクト中の bond オブジェクトを全て取り出す。
[bond for bond in mol.GetBonds()]
Bond オブジェクトの結合次数を取り出す。共役系は 1.5 となる。
bond.GetBondTypeAsDouble()
Bond オブジェクトの結合を構成する原子の atom オブジェクトを取り出す。結合を構成する原子は 2 つあるのでそれぞれ別の関数で取り出す。
bond.GetBeginAtom() bond.GetEndAtom()
分子中の部分構造を検索。出力は部分構造に対応する原子の index のタプル。複数ある場合、index が最も若い一種類だけが返ってくる。反対に、部分構造がヒットしなければ空のタプルが返ってくる。
mol = Chem.MolFromSmiles("c1c(Cl)c(c2nccc(OCCC)c2)ccc1C(=O)NC1CC1") query = Chem.MolFromSmiles("CCC") mol.GetSubstructMatch(query)
検索した部分構造に対応する原子の index の組み合わせ全てを返す。
mol.GetSubstructMatches(query)
Distance geometry 法による立体配座の発生。
from rdkit.Chem.rdDistGeom import ETKDGv3, EmbedMolecule, EmbedMultipleConfs # Distance geometry で立体配座を生成 param = ETKDGv3() # Seed 値は 0 だと固定されない param.randomSeed = 1 # 立体廃座が生成できていれば 0 を返す。入力した mol オブジェクトは立体配座情報が付与される。入力する mol オブジェクトに水素原子を付加することを忘れずに。 EmbedMolecule(mol_h, param)
生成した立体配座の座標を表示。
mol_h.GetConformer().GetPositions()
複数の立体配座をまとめて生成。
from rdkit.Chem.rdDistGeom import EmbedMultipleConfs param = ETKDGv3() # 10 個の立体配座を生成。mol_h に配座情報が加わるので、mol オブジェクトのリストみたいにはならない。 EmbedMultipleConfs(mol_h, 10, param)
立体配座のリストを表示
list(mol_h.GetConformers())
Mol オブジェクトを sdf として保存する。
mols = [Chem.MolFromSmiles(smiles) for smiles in ["CCO", "c1ccccc1", "N[C@@H](C)C(=O)O"]] writer = Chem.SDWriter("saved_mols.sdf") for mol in mols: writer.write(mol) writer.close()
SDF の読み込み
supplier = Chem.SDMolSupplier("saved_mols.sdf") # Supplier は mol オブジェクトのリストに近い操作ができる。 supplier[0]
小ネタ:Fingerprint の算出方法
RDKit にはさまざまな fingerprint が実装されています。
おそらく最もよく使われている fingerprint は Morgan fingerprint (ECFP)ですが、他にも MACCS Key や SMILES extended-connectivity fingerprint (SECFP)、MinHash fingerprint (MHFP) などの計算も可能です。
MHFP と SECFP については以下の Reymond の原著論文を参照して下さい。
RDKit で各 fingerprint を計算するサンプルコードは以下の通りです。
from rdkit.Chem.rdMolDescriptors import GetMACCSKeysFingerprint, GetMorganFingerprintAsBitVect from rdkit.Chem.rdMHFPFingerprint import MHFPEncoder # Mol オブジェクトのリストを用意 mols = [Chem.MolFromSmiles(smiles) for smiles in ["CCO", "c1ccccc1", "CC(=O)Oc1ccccc1C(=O)O"]] # MACCS Key maccs = np.array([GetMACCSKeysFingerprint(mol) for mol in mols]) # ECFP4 ecfp4 = np.array([GetMorganFingerprintAsBitVect(mol, 2, 2048) for mol in mols]) # ECFP6 ecfp6 = np.array([GetMorganFingerprintAsBitVect(mol, 3, 2048) for mol in mols]) # FCFP4 ecfp4 = np.array([GetMorganFingerprintAsBitVect(mol, 2, 2048, useFeatures = True) for mol in mols]) # FCFP6 ecfp4 = np.array([GetMorganFingerprintAsBitVect(mol, 3, 2048, useFeatures = True) for mol in mols]) # SECFP4 secfp4 = np.array([MHFPEncoder().EncodeSECFPMol(mol, 2) for mol in mols]) # SECFP6 secfp4 = np.array([MHFPEncoder().EncodeSECFPMol(mol, 3) for mol in mols]) # MHFP4、binary vector ではなく、整数のベクトルが得られる。 mhfp4 = np.array([MHFPEncoder().EncodeMol(mol, 2) for mol in mols]) # MHFP6、binary vector ではなく、整数のベクトルが得られる。 mhfp6 = np.array([MHFPEncoder().EncodeMol(mol, 3) for mol in mols])
MACCS Key は 167 次元のベクトルが出力されますが、最初の次元は必ず 0 なので、それを除いた 166 次元のベクトルを解析に用いて下さい。
各 fingerprint の数字は fingerprint に対応する fragment の直径のパスの長さを示します。関数の引数は半径のパスの長さなので、注意して下さい。
MHFP は Reymond のグループが公開しているものと挙動が異なる可能性があります(要検証)。
算出した fingerprint は保存することもできます。
以下に fingerprint の保存例を示します。
# Fingerprint の算出 mol = Chem.MolFromSmiles("CC(=O)Oc1ccccc1C(=O)O") fp = GetMorganFingerprintAsBitVect(mol, 2, 2048) # Fingerprint を文字列表記する fp_str = fp.ToBase64() # テキストファイルなどにして保存 with open("ecfp4.txt", "a") as writer: writer.write()
Fingerprint の復元例は以下の通りです。
from rdkit.DataStructs.cDataStructs import ExplicitBitVect # テキストファイルから fingerprint の文字列を読み込む with open("ecfp4.txt", "r") as reader: fp_str = reader.read() # Fingerprint 復元用のインスタンスを作成。Vector の次元数を引数とする re_fp = ExplicitBitVect(2048) # Fingerprint の文字列を読み込ませて復元する re_fp.FromBase64(fp_str) # 復元した fingerprint が元の fingerprint と一致しているか確認 re_fp == fp
ご注文は国際学会ですか? ~にわのフランス珍道中~
###注意###
今回の記事は、新型コロナウイルス、ウクライナ戦争、政治に関する言及があります。全て私個人の感想であり、いかなる組織、国家、集団等の意見を代弁するものではありません。本記事閲覧の際は、上述の点に御留意下さい。
はじめに
Bonjour!
ついこの間、国際学会に現地で発表をしてきました!
会場が何とフランスのストラスブール!ストラスブールといえばお洒落な街並みで知られている EU 議会のある都市であり、アルザスワインの聖地であり、ごちうさの聖地であり、そして何より、化学の聖地です!昔からストラスブールに行きたい、と思っていたので、学会がオンサイトで開かれると聞いて、無理をしてでも行くことにしました。
今回はケモインフォマティクスの話はほぼせず、浮かれポンチの旅行日記フランスの学会参加にあたり感じたことを記して行こうと思います。
フランスに行く準備(2022 年 7 月 1 日時点)
大前提として、ワクチン 3 回接種が事実上必要です。指定されたワクチン(ファイザー、モデルナいずれでも OK)を 3 回接種していれば、フランスの入国審査で止められることはありません。ワクチンパスポートも取得しておきましょう。反対に、日本に帰国するのが大変で、出国 72 時間前の PCR 検査結果が求められます。私はシャルルドゴール空港(パリ近くの一番大きい空港)の PCR センターを予約しましたが、手続きが全部フランス語で書かれており、英語の書類が一切ないので、めちゃくちゃ大変です。PCR 検査は、値段は 70 ユーロしますが、rapid PCR がオススメです。検査してから 1 時間せずに結果が帰ってきました。間違っても antigen test (fast test) は選択しないで下さい!日本入国の書類として認められていません。後、コロナに感染した場合の保証がある海外旅行保険の加入も必須です。フランスの PCR 検査で陽性となったらその瞬間、冗談ではなく本当に日本に帰れなくなります。PCR で陽性になった場合、どう生き残るかはシミュレートしておいた方がいいです。日本に入国する際、MySOS というアプリをスマホにインストールしておくと、日本の入国は楽です。MySOS の操作はかなり楽です。
飛行機のチケットは、フランスに行くと決めたら極力早く確保した方が良いです。基本高い上に、日本-海外間の飛行機の本数が少なくなっているので、エコノミーが埋まっている、ということはざらにあります。さらに、頭おかしいレベルでトランジットする羽目になるので、チケット確保は極力早くがポイントです。私はエティハド航空というアラブ首長国連邦の航空会社を利用しました。
後は着替えぐらいで特別なものは持っていきませんでしたが、シャンプーや石鹸、歯ブラシなどは持っていった方が良いです。フランスのホテルには日本で当たり前にあるアメニティがないことも少なからずあるので。
ストラスブールまでの道のり
成田からアラブ首長国連邦のアブダビ空港を経由して、パリのシャルルドゴール空港に向かいました。成田空港はドン引きするぐらいガラガラで、日本の入出国がいかに少ないかを感じました。海外出張自体が人生初だったからか、飛行機の座席が硬かったからか、普段睡眠時間が長い人間としてはかなり短い時間(3 時間ぐらい?)しか眠れませんでした。機内食は思ったよりいけました。アブダビ空港ではマスクをしている人も少なくなり(1 割ぐらい?)ましたが、歩いている人の人種が多様であり、フランスに着く前に日本とは違うというのを感じました。
フランス行きの飛行機に乗ると、隣の人が話しかけてきました。何でもオーストラリアの大学で数理物理学を研究している研究者で、フランスの別の都市で開催される学会に参加するようです。私はゴミみたいな英語(以下ゴミングリッシュ)しか話せないのですが、それでもそれなりに会話は楽しみました。
シャルルドゴール空港に着くと、地下鉄に乗ってパリの中心部へ向かいました。パリの地下鉄は治安が悪いことで有名で、スリも多いと聞いていたので、セキュリティバッグに貴重品を入れて、肌身離さず持ち歩きました。幸いスリには出くわしませんでしたが、日本では考えられないレベルであちこちに落書きがあり、床も汚かったので、日本が清潔、安全な国であるとも感じました。
フランスには学会前日入りしているので、少しパリを観光しました。あくまで出張の空き時間にですよ。
有名どころだけですが、凱旋門に行ってきました。中央のアーチの真下は、第一次大戦の慰霊碑になっていました。エッフェル塔は、近くまで行きました。建物一つ一つがお洒落で、現実味が湧きませんでした。ただし、日本よりもホームレスは優位にたくさん見かけましたが・・・
時差ボケを治すため宿で少し昼寝してから夕食を取りました。宿の目の前のレストランで食事をしましたが、20 ユーロでサラダ + ムニエル + デザート + ワインなので、実はそこまで外食は極端に高くないのでは?と思いました。量も多いですし。何よりデザートのフォンダンショコラが洒落にならないぐらい美味しい!今回のフランス滞在で一番美味しかったかもしれません。この後も色んなスイーツを食べましたが、何一つはずれがありませんでした。さすが美食の国。
グラフ畳み込みニューラルネットワークによる回帰モデル:Deep Graph Library(DGL)の使い方
グラフ畳み込みニューラルネットワークのコードの例です。
R2 は -0.05 ぐらいと低いですが、計算自体は行えます。
(解説は近日追記)
import numpy as np import pandas as pd import torch import torch.nn as nn import torch.nn.functional as F from torch.optim import Adam import dgl from dgl.dataloading import GraphDataLoader from dgl.nn import GraphConv from rdkit import Chem from rdkit.Chem.rdPartialCharges import ComputeGasteigerCharges from rdkit.Chem.Draw import IPythonConsole, MolsToGridImage from sklearn.model_selection import train_test_split from sklearn.metrics import r2_score # 化学構造の読み込み structure = Chem.SDMolSupplier("../data/logSdataset1290_2d.sdf") # データをランダムに分割 np.random.seed(0) tag_list = list(range(len(structure))) np.random.shuffle(tag_list) train_tag = np.array(tag_list)[:1200] test_tag = np.array(tag_list)[1200:] train_mols = [] test_mols = [] train_y = [] test_y = [] for i in train_tag: mol = structure[int(i)] train_mols.append(mol) train_y.append(np.float(mol.GetProp("logS"))) for i in test_tag: mol = structure[int(i)] test_mols.append(mol) test_y.append(np.float(mol.GetProp("logS"))) # ノード単位で記述子を算出する関数 def get_node_features(mol): node_features = [] for atom in mol.GetAtoms(): node_features.append([atom.GetAtomicNum(), np.int(atom.GetIsAromatic()), np.float(atom.GetProp("_GasteigerCharge"))]) return torch.from_numpy(np.array(node_features)).float() # ノードの情報を取り出す関数 def get_graph_data(mols): graphs = [] for mol in mols: # Gasteiger charge を計算 ComputeGasteigerCharges(mol) # 結合しているノードの組み合わせを算出 node_pair = np.where(np.triu(Chem.GetAdjacencyMatrix(mol)) == 1) # Graph 型のデータを作成 graph = dgl.graph((node_pair[0], node_pair[1]), num_nodes = len(mol.GetAtoms())) # ノード毎の特徴量を入力 graph.ndata["atom_features"] = get_node_features(mol) # 自己ノードを畳み込むための self loop を設定する graph = dgl.add_self_loop(graph) graphs.append(graph) return graphs # 化学構造(Mol オブジェクト)から Graph 型のデータを作成 train_graph = get_graph_data(train_mols) test_graph = get_graph_data(test_mols) # 目的変数の読み込み train_y_tensor = torch.tensor(train_y).float() test_y_tensor = torch.tensor(test_y).float() # Data loadder の定義 train_dataloader_input = list(zip(train_graph, train_y_tensor)) train_loader = GraphDataLoader(train_dataloader_input, batch_size = 64, drop_last = False, shuffle = True) test_dataloader_input = list(zip(test_graph, test_y_tensor)) test_loader = GraphDataLoader(test_dataloader_input, batch_size = 64, drop_last = False, shuffle = False) # モデルのクラスを設定 class Model(nn.Module): def __init__(self): super().__init__() self.conv_1 = GraphConv(3, 8) self.conv_2 = GraphConv(8, 16) self.linear_1 = nn.Linear(16, 8) self.norm = nn.BatchNorm1d(8) self.linear_2 = nn.Linear(8, 1) def forward(self, g, f): h = F.relu(self.conv_1(g, f)) h = self.conv_2(g, h) g.ndata["hidden"] = h x = dgl.mean_nodes(g, "hidden") x = F.relu(self.linear_1(x)) x = self.norm(x) x = self.linear_2(x) return x # モデルのインスタンスを作成 model = Model() # 最適化関数の設定 optimizer = Adam(model.parameters(), lr = 0.001) # 損失関数の設定 loss_function = nn.MSELoss() # モデルの学習 epoch = 30 model.train() for i in list(range(epoch)): epoch_loss = 0 # バッチごとの処理 for batch_graph, batch_y in train_loader: # トレーニングデータに対する誤差の算出 batch_pred_y = model(batch_graph, batch_graph.ndata["atom_features"]) batch_loss = loss_function(batch_pred_y, batch_y) # 誤差逆伝播と重み更新 optimizer.zero_grad() batch_loss.backward() optimizer.step() epoch_loss += batch_loss.item() # エポックごとの誤差を表示 print(epoch_loss) # モデルの推論 model.eval() with torch.no_grad(): test_loss = 0 pred_y = torch.Tensor() # トレーニングと同様の処理を行うが、重みの更新はしない for batch_graph, batch_y in test_loader: batch_pred_y = model(batch_graph, batch_graph.ndata["atom_features"]) batch_loss = loss_function(batch_pred_y, batch_y) test_loss += batch_loss.item() pred_y = torch.cat([pred_y, batch_pred_y]) # 誤差を表示 print(test_loss) # Numpy 型に戻した後に R2 を算出 pred_y = pred_y.detach().numpy() print(r2_score(test_y, pred_y))
とりあえず PyTorch: 最低限これで動く
ニューラルネットワーク(深層学習)ブームは 2022 年現在も続いており、毎日新しい研究がニュースや SNS で話題となっています。ケモインフォマティクス分野でも例外ではなく、QSAR モデルや構造生成器、逆合成解析を始めとして様々な深層学習を用いた研究がされています。
深層学習のライブラリは Tensorflow や Caffe なども知られていますが、この記事を執筆している時点では、PyTorch が盛んに使われています。しかし、PyTorch でとりあえず QSAR モデルを組みたい、という場合に、意外と日本語のページがヒットしないような気がします。
そこで、非常に最低限ですが、とりあえず回帰モデルを組みたい、という場合に使える基本的なコードを作成してみました。
import numpy as np import pandas as pd import torch import torch.nn as nn import torch.nn.functional as F from torch.utils.data import TensorDataset, DataLoader from torch.optim import Adam from sklearn.metrics import r2_score # データの読み込み train_X = pd.read_csv("../data/LogS_train_scaled.csv", index_col = 0) test_X = pd.read_csv("../data/LogS_test_scaled.csv", index_col = 0) train_y = pd.read_csv("../data/LogS_train_value.csv", index_col = 0) test_y = pd.read_csv("../data/LogS_test_value.csv", index_col = 0) # データを Tensor 型にする train_X_tensor = torch.Tensor(np.array(train_X)).float() train_y_tensor = torch.Tensor(np.array(train_y)).float() train_tensor = TensorDataset(train_X_tensor, train_y_tensor) test_X_tensor = torch.Tensor(np.array(test_X)).float() test_y_tensor = torch.Tensor(np.array(test_y)).float() test_tensor = TensorDataset(test_X_tensor, test_y_tensor) # Data loader を作成 train_loader = DataLoader(train_tensor, batch_size = 32, shuffle = True) test_loader = DataLoader(test_tensor, batch_size = 32, shuffle = False) # モデルのクラスを設定 class Model(nn.Module): def __init__(self): super().__init__() self.linear_1 = nn.Linear(94, 32) self.linear_2 = nn.Linear(32, 1) self.norm = nn.BatchNorm1d(32) def forward(self, x): x = F.relu(self.linear_1(x)) x = self.norm(x) x = self.linear_2(x) return x # モデルのインスタンスを作成 model = Model() # 最適化関数の設定 optimizer = Adam(model.parameters()) # 損失関数の設定 loss_function = nn.MSELoss() # モデルの学習 epoch = 20 model.train() for i in list(range(epoch)): epoch_loss = 0 # バッチごとの処理 for batch_X, batch_y in train_loader: # トレーニングデータに対する誤差の算出 batch_pred_y = model(batch_X) batch_loss = loss_function(batch_pred_y, batch_y) # 誤差逆伝播と重み更新 optimizer.zero_grad() batch_loss.backward() optimizer.step() epoch_loss += batch_loss.item() # エポックごとの誤差を表示 print(epoch_loss) # モデルの推論 model.eval() with torch.no_grad(): test_loss = 0 pred_y = torch.Tensor() # トレーニングと同様の処理を行うが、重みの更新はしない for batch_X, batch_y in test_loader: batch_pred_y = model(batch_X) batch_loss = loss_function(batch_pred_y, batch_y) test_loss += batch_loss.item() pred_y = torch.cat([pred_y, batch_pred_y]) # 誤差を表示 print(test_loss) # Numpy 型に戻した後に R2 を算出 pred_y = pred_y.detach().numpy() print(r2_score(test_y, pred_y)) # 構築したモデルの保存 torch.save(model.state_dict(), "../model/nn_model.pth")
今回は、94 次元の記述子から、pLogS(連続値)を予測するモデルとなっています。データサイズ はトレーニングデータ 1,200、テストデータ 94 なのでとても小さいです。大体の目安ですが、10,000 データ以上の場合、深層学習は力を発揮します。
最初のポイントは、データを Tensor 型にするところです。PyTorch は基本的にデータを Tensor 型にしないと計算してくれないので、Numpy のフレームから変更する必要があります。また、PyTorch では int 型や float64 型などは学習に用いることができないので、float() で float32 型にしてあげます。説明変数(X)と目的変数(y)を一つの DataFrame にまとめてから、バッチを作成する関数である DataLoader に入力します。ここでバッチサイズを決めます。バッチサイズは大雑把ですが、データ数にルートを取った値ぐらいを試すのがちょうど良い気がします(データ数が数マンだったら 256 ぐらい、といった感じ)。
深層学習モデルのクラスは、 __init__ と forward から構成されます。__init__ にモデルで使う全結合層や畳み込み層などを定義します。__init__ に super などがついている場合もありますが、少し古い書き方だそうで、基本的に今回の例のようなシンプルな書き方で問題ありません。forward で関数を組み合わせて、モデルの形状を定義します。今回は batch normalization と活性化関数(ReLU)を挟んだ、二層の全結合層モデルとなっています。データサイズが少ない(20,000 以下ぐらい?)場合は、モデルを複雑にしても予測精度は向上しない気がします。今回は回帰問題なので出力層はそのままですが、判別問題の場合は、各クラスの確率を出力したいので、ソフトマックス関数を追加することを忘れないようにしましょう。
モデルのインスタンスと最適化関数、損失関数を定義します。最適化関数は SGD でも良いのですが、とりあえずよく使われている Adam を今回は採用します。model.parameters() はおまじないとして、入力を忘れないようにして下さい。学習率(lr)は、データサイズが大きい場合に小さくすると良いです。1e-4 をとりあえず試して見るのが良いかもしれません。損失関数は平均二乗誤差を用います。判別問題の場合はクロスエントロピーを用いて下さい。
いよいよモデルの学習です。エポック数は本当は early stopping を実装して決めるべきなのですが、今回は適当に 20 に設定します。定義した DataLoader から for 文でデータを取り出します。モデルのインスタンスにデータを入力して予測値を算出し、誤差を算出します。その後に、誤差逆伝播法を実行します。勾配の初期化、誤差逆伝播の具体的な計算、最適化関数のパラメータ更新などを行っているそうですが、ここもまずはおまじないということで良いでしょう。
モデルの学習が出来たら、推論を行います。折角学習した重みに変更を加えたくないので、model.eval() と torch.no_grad() を両方実行します。計算方法は学習時と基本的に同じですが、誤差逆伝播法は当然行いません。予測値を、torch.cat を使って保存していきます。
出力値の R2 を計算してみます。Tensor 型から NumPy 型に変換しますが、勾配の学習に用いた Tensor は、一度 detach() を挟む必要があります。とりあえずつけてから numpy() を実行するようにしておくとエラーが起きにくいと思います。
シード値をつけていないので、多少ズレはあるのですが、R2 は大体 0.88 前後と、適当に組んだ割にはまあまあ良い値となりました。
保存したモデルの読み込みは以下の通りです。
# モデルの読み込み loaded_model = Model() loaded_model.load_state_dict(torch.load("../model/nn_model.pth"))
モデル構築に用いたクラスを読み込み、そこに学習したモデルの重みを入れるイメージです。
今回のコードは CPU で実行しました。GPU に対応させる場合は、バッチのデータとモデルに cpu() を追加して下さい。また、用いる GPU のバージョンに対応した PyTorch が適切にインストールされているかも注意点です。
最低限の内容ですが、とりあえず動かしたい、という場合の参考になれば幸いです。
決定木で AD を算出:Isolation forest
予測モデルを扱う際に、applicability domain (AD) を考慮することは重要です。
AD を算出する方法としては k 最近傍法や one-class SVM、アンサンブル法などが代表的です。
決定木と言えば Light GBM や XGBoost、ランダムフォレストなどでお馴染みですが、異常検知手法として用いられる決定木ベースの手法も存在します。
それが isolation forest であり、scikit-learn で扱うことが可能です。
import numpy as np import pandas as pd from sklearn.ensemble import IsolationForest # データの読み込み data = np.array(pd.read_csv("../data/logSmols_rdkit_200.csv", index_col = 0)) # モデルを構築 model = IsolationForest(contamination = 0.2, random_state = 0) ad_label = model.fit_predict(data)
モデル構築はいつもの scikit-learn の枠組みでできるので非常に楽です。引数の contamination が異常値の割合を示します。ランダムフォレストや Light GBM などと違い、feature_importances_ で寄与率を確認することはできませんが、モデル中の各決定木をリストを model.estimators_ で表示させ、そこから決定木を選択すると、対応する feature_importances_ を出すことができます。
fit_predict の返り値は OCSVM と同様に 1 or -1 であり、-1 だと異常値、即ち AD 外とみなすことができます。
model.decision_function(data) を使うと、予測結果として連続値を得ることができます。値が正だと fit_predict の 1 に、値が負だと fit_predict の -1 にそれぞれ対応します。
Isolation forest の結果が妥当そうかどうか、PCA plot を用いて確認してみましょう。
import matplotlib.pyplot as plt from sklearn.decomposition import PCA from sklearn.preprocessing import StandardScaler # データのオートスケーリング scaler = StandardScaler() data_scaled = scaler.fit_transform(data) # 分散が 0 (全て同じ値)の記述子は除去 data_scaled = data_scaled[:, np.std(data_scaled, 0) != 0] # PCA を実施 pca = PCA() data_pca = pca.fit_transform(data_scaled) # フォントを変更 plt.rcParams["font.family"] = "Arial" # Plot のインスタンスと図の大きさを定義 fig = plt.figure(figsize = (10, 10)) # インスタンスの中に複数のグラフを作る場合に指定 # 引数は(行数、列数、具体的に作りたい図のインデックス) # グラフを 1 つだけ作りたいときは (1, 1, 1)としてやれば良い ax = fig.add_subplot(1, 1, 1) # グラフのタイトル ax.set_title("Isolation Forest AD", fontsize = 28) # グラフの表示範囲 ax.set_xlim(-10, 35) ax.set_ylim(-15, 20) # 目盛の数字の大きさを調整 ax.tick_params(axis = "x", labelsize = 16) ax.tick_params(axis = "y", labelsize = 16) # 軸のラベル名 ax.set_xlabel("PC1", fontsize = 20) ax.set_ylabel("PC2", fontsize = 20) # データをプロットする ax.scatter(data_pca[ad_label == 1, 0], data_pca[ad_label == 1, 1], c = "dodgerblue", label = "in AD") ax.scatter(data_pca[ad_label == -1, 0], data_pca[ad_label == -1, 1], c = "crimson", label = "out AD") # 凡例を表示 plt.legend() # 図を保存 fig.savefig("../result/Isolation_forest_ad.png")
得られたプロットは以下の通りです。
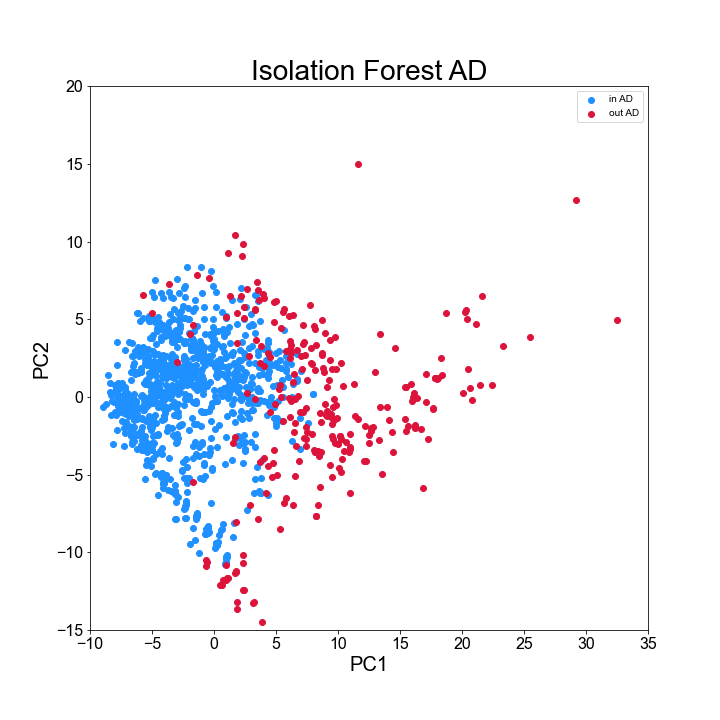
赤が AD 外と判断されたデータですが、プロットの端に位置する傾向が見られ、結果は妥当そうだと考えることができます。
小ネタ:RDKit による分子記述子の算出
RDKit はケモインフォマティクスをやるのに欠かせないライブラリです。
分子の描写など、色々できますが、分子記述子を算出することもできます。
ちなみに、Mordred も、RDKit をベースに記述子を計算しています。
RDKit で分子記述子を算出するコードは以下の通りです。
import numpy as np import pandas as pd from rdkit import Chem from rdkit.Chem import Descriptors from rdkit.ML.Descriptors.MoleculeDescriptors import MolecularDescriptorCalculator # 化学構造データの読み込み supplier = Chem.SDMolSupplier("../data/logSdataset1290_2d.sdf") smiles_list = [Chem.MolToSmiles(mol) for mol in supplier] # 記述子名の抽出 descriptor_names = [descriptor_name[0] for descriptor_name in Descriptors._descList] # 記述子を計算するインスタンスの作成 calculator = MolecularDescriptorCalculator(descriptor_names) # 記述子計算 descriptors = [calculator.CalcDescriptors(mol) for mol in supplier] # DataFrame 形式にする descriptors = pd.DataFrame(descriptors, index = smiles_list, columns = descriptor_names)
上記のコードは、以下の記事を参考にさせて頂きました。
何をしているか少し解説をすると、MolecularDescriptorCalculator が記述子を計算してくれるインスタンスで、(RDKit に対応している)記述子名を入れると、その記述子を計算してくれます。実は、Descriptors._descList は記述子名とその記述子を計算してくれる関数のタプルが集合しているインスタンスなので、特定の記述子だけ計算したい場合は、ここから上手く選択してやるのも良いかもしれません。
インスタンスである calculator から記述子名の一覧を取得したいときは、calculator.GetDescriptorNames() を使うと便利です。また、記述子の簡単な説明が欲しい場合は、calculator.GetDescriptorSummaries() を使うと、記述子の説明や参考文献が出てきます(N/A となっている記述子もあるので注意)。
2022 年 1 月現在、上記の方法で 200 種類の記述子を取得することが可能です。
余談ですが、CompoundDescriptors というクラスもあり、公式の説明をざっと読んだ感じ、原子レベルの記述子を MoleculeDescriptors と同じ要領で計算できそうな雰囲気がするのですが、記述子名の取得が分からず断念・・・
今回の記事も自分のメモ用ですが、誰かの調べる手間が少しでも軽減すれば幸いです。